The Use of Platform Methods for Process Related Impurities in Biologics

Developing validated analytical methods for every process-related impurity in a biologics program is resource-intensive. Drawing on deep experience in biologics analytical testing, Element expert Khanh Courtney examines how platform methods, adapted to new products through matrix feasibility studies, can compress development timelines, and when custom development is the right approach.
Biological product development requires comprehensive characterization of process-related impurities at every stage, from early research through commercial manufacturing. For each identified critical quality attribute, sponsors need validated analytical methods that can accurately detect and quantify potential contaminants. The traditional approach involves developing custom analytical methods for each impurity in each product. This works but creates sequential dependencies that can extend development timelines significantly.
Platform methods offer an alternative worth considering. These are standardized, pre-developed analytical procedures designed to detect common process-related impurities across multiple biologics modalities and formulations. Rather than starting from scratch each time, laboratories can adapt existing methods to new products through matrix feasibility studies.
This article examines how platform methods function within biologics analytical development, when they make strategic sense, and how to determine if they're appropriate for your program.
Understanding Process-Related Impurities in Biologics Manufacturing
Product-related impurities are molecular or post-translational variants that arise during manufacturing or storage. These might include degradation products, aggregates, deamidation, oxidation, or other molecular modifications that alter the therapeutic properties of the drug substance. While related to the product itself, these variants don't share the same activity, efficacy, or safety profile as the intended molecule.
Process-related impurities come from the manufacturing process itself. They originate from three primary sources: the cell substrate (host cell proteins and residual DNA), the cell culture process (media components, antibiotics, inducers), and downstream processing (chromatography ligands, processing enzymes, chemical reagents, and packaging). Each manufacturing step introduces potential contaminants that need to be monitored and controlled.
The U.S. Food and Drug Administration defines critical quality attributes as properties or characteristics that must remain within appropriate limits to ensure desired drug quality and safety. Process-related impurities are usually associated with safety. When an impurity is identified as a CQA, it requires analytical methods to ensure appropriate levels of clearance from the drug substance or drug product. The challenge lies in developing these methods efficiently without compromising scientific rigor.
Why Process-Related Impurities Control Matters for Biologics Manufacturing
Impurities in biological products aren't just a regulatory checkbox. Some present direct safety risks through immunogenicity or toxicity. Others may affect product stability, compromising efficacy over the shelf life. Even impurities without immediate safety concerns can impact the overall quality profile.
Demonstrating effective impurities control requires more than just testing the final product. Development programs need to show they understand where impurities come from, how processing steps clear them, and what levels remain acceptable. This means establishing robust analytical methods early enough to support process development, then maintaining those methods through process performance qualification and commercial manufacturing.
The analytical strategy needs to evolve as programs advance. Early development might rely on screening-level methods to compare process options. Later stages might require fully validated procedures that meet regulatory expectations for CMC testing. The question becomes how to build this analytical capability efficiently while maintaining the flexibility to adapt as programs progress.
What Are Platform Methods for Process-Related Impurities Testing?
Platform methods represent a different approach to analytical method development. Instead of creating unique procedures for each product, laboratories develop robust methods for common process-related impurities that can be adapted across multiple programs. The methods are designed with enough flexibility to accommodate different product matrices while maintaining expected performance.
Consider host cell protein testing as an example. Most biologics manufacturing involves removing host cell proteins from the cell substrate used for production. Whether you're producing a monoclonal antibody or a recombinant protein, you'll need to quantify residual host cell proteins and show clearance. A platform ELISA method developed for CHO cell proteins can potentially serve multiple CHO-expressed products with appropriate performance parameters for each specific matrix.
The same principle applies to other common process-related impurities. Protein A leachables from affinity chromatography, residual DNA from host cells, processing enzymes like Benzonase, and purification reagents like CsCl appear across many biologics manufacturing processes. Platform methods for these impurities can be applied to new products more quickly than developing custom procedures from the beginning.
This doesn't mean platform methods work for everything. Product-specific impurities, protein concentration, harsh formulation buffers containing high salt or sugar concentrations, or novel processing reagents may require custom method development. The strategic question is identifying where platform methods apply and where custom development is necessary.
When to Use Platform Methods vs Custom Analytical Development
Not every analytical challenge calls for a platform approach. The decision depends on several factors: the impurity being measured, the product matrix, the development stage, and the regulatory strategy.
Platform methods work best for well-characterized impurities with established detection approaches. If an impurity has been measured across many products using similar analytical techniques, there's likely a platform method that can be adapted. Conversely, novel impurities or those requiring cutting-edge analytical approaches may need custom development regardless of timeline pressures.
The product matrix matters significantly. Biologics formulations vary widely in protein concentration, buffer composition, excipients, and other components that can interfere with analytical methods. A platform method that works beautifully in one formulation might fail in another due to matrix effects. Matrix feasibility testing evaluates whether a platform method performs adequately in your specific product, but high protein or salt concentration, or unusual formulations may present challenges.
The development stage influences the decision as well. Early programs often benefit most from platform methods because speed matters more than ultimate optimization. Getting reliable data quickly helps guide process development decisions. Later-stage programs heading toward regulatory submissions need thoroughly validated methods, but even then, starting with a proven platform can accelerate the validation timeline compared to developing methods from scratch.
Your regulatory strategy also plays a role. If you're pursuing an accelerated pathway or have aggressive clinical timelines, platform methods can help you meet milestones without sacrificing data quality. For programs with more flexible timelines, the decision becomes more about resource allocation and technical preferences.
Platform Method Decision Framework for Biologics Programs
The first question is whether the impurity qualifies as a safety risk that requires formal assessment. Not every potential contaminant needs validated testing, and focusing resources on actual safety risk factors, including toxicity levels, makes strategic sense. This evaluation and risk assessment should be performed by the sponsor.
For impurities requiring assessment, the next question is whether a suitable platform method exists. Element maintains established procedures for many common process-related impurities found in the biologics manufacturing process. If a relevant platform method exists, matrix feasibility testing can quickly establish whether it performs adequately in the specific product matrix.
Matrix feasibility studies evaluate several performance parameters. Specificity testing confirms the method detects the target impurity without interference from the drug substance or formulation components. Accuracy assessment through spike recovery experiments verifies that the method can accurately measure the impurity at relevant concentrations, such as detection and quantitation limits. Precision evaluation demonstrates the method produces consistent results. These studies typically require less time than full method development because the analytical procedure itself is already established.
If matrix feasibility demonstrates acceptable performance, the platform method can be used for sample analysis. For non-GMP early development work, this might be sufficient. For regulatory submissions, additional validation may be required, but starting with a proven platform method provides a solid foundation that reduces validation timelines.
When no suitable platform method exists, or when method performance is poor in the matrix of a specific purification step, it is possible to consider assessing a potentially “friendlier” matrix further downstream. If that is not possible, method optimization or custom method development becomes necessary. Custom development takes longer but sometimes represents the only viable path forward for challenging impurities or unusual matrices.
The framework isn't about always choosing platform methods or always developing custom procedures. It's about making informed decisions based on technical requirements, timeline constraints, and resource availability.
Common Process-Related Impurities in Biologics Manufacturing
Certain impurities appear frequently enough in biologics manufacturing that platform methods have become well-established. Understanding which impurities fall into this category helps in planning analytical strategies.
Antibiotics used for cell selection during early fermentation stages need quantification to ensure adequate clearance. Kanamycin, streptomycin, and other aminoglycoside antibiotics have standardized testing procedures that can be adapted to various product matrices. These small molecules are relatively straightforward to measure using liquid chromatography-mass spectrometry techniques.
Expression inducers like IPTG (isopropyl β-D-1-thiogalactopyranoside) are commonly used in bacterial expression systems. Platform methods exist for IPTG and similar compounds, allowing rapid assessment of residual levels in purified drug substance.
Processing enzymes like Benzonase (used for nuclease digestion) or Trypsin, and growth factors used in upstream mammalian cell culture media need quantification. These protein-based impurities can be measured using platform immunoassays or activity-based methods, depending on the specific enzyme.
Host cell proteins and residual DNA represent universal concerns across biologics production. While HCP ELISAs are often product-specific or process-specific, some platform assays exist for common expression systems like CHO cells or E. coli. Residual DNA quantification by ddPCR represents a well-established platform approach applicable to most biologics.
Host Cell Protein and Residual DNA: Key Process-Related Impurities
Host cell protein testing deserves special attention because it represents one of the most critical process-related impurities for a biologics program. HCP contamination can trigger immunogenic responses in patients, making thorough characterization and control essential for product safety.
Platform HCP ELISA methods exist for common expression systems, particularly Chinese Hamster Ovary (CHO) or Human Embryonic Kidney (HEK) cells. These assays use polyclonal antibodies generated against the host cell proteome to detect a broad range of potential HCP contaminants. While some programs require process-specific HCP ELISAs, generic platform assays can provide adequate coverage for early development and can inform decisions about whether custom assay development is necessary.
Residual DNA testing similarly benefits from platform approaches. Quantitative PCR methods such as ddPCR can detect host cell DNA at the low levels required by regulatory guidance. These methods work across different host cell types with appropriate reference standards and validation.
Both HCP and residual DNA testing demonstrate the value of platform methods in biologics analytical development. Rather than developing these assays from scratch for each program, laboratories can leverage established procedures and focus custom development efforts on truly unique analytical challenges.
Implementing Platform Methods in CMC Programs
Applying platform methods for residual process impurities effectively requires a safety risk assessment of the manufacturing process, including the input, intermediates, and the final output. This involves a review of raw materials, ancillary reagents, expression systems, purification schemes, and formulation components to identify potential impurities requiring assessment. While this article does not address extractables and leachables, they must be considered especially at a later stage in the development of the product.
For impurities with suitable platform methods, matrix feasibility may commence as soon as the samples are received. The intent of the matrix feasibility assessment is to determine if the platform method is suitable for use with the client sample matrices. Sample matrices may represent the final drug substance, or samples taken from intermediate steps in the purification process. Specificity, Accuracy by spike/recovery at the LOD and LOQ levels, and precision assess the performance of the method in the client-provided sample matrices.
If matrix feasibility confirms adequate performance, sample analysis can begin immediately for early phase, or process development samples. For early development work, matrix feasibility alone may suffice. For regulatory submissions, validation of the method will be required for the test sample types. Even with additional validation, starting from an established base platform method provides significant advantages over beginning with an entirely new procedure.
When matrix feasibility reveals issues due to matrix effects, the approach shifts. Sometimes, minor method modifications can address the problem. Other times, custom method development becomes necessary. The matrix feasibility testing itself provides valuable information about what's causing the problem, which accelerates custom development if needed.
When Custom Development Is Necessary for Biologics
While platform methods provide advantages for many common process-related impurities, certain situations require custom analytical development. Novel or advanced biologics modalities, such as cell and gene therapies, may involve unique impurities or specific growth factors used in the upstream cell culturing process. Platform methods may not exist for specialized programs utilizing novel or non-traditional manufacturing processes with modality-specific impurities.
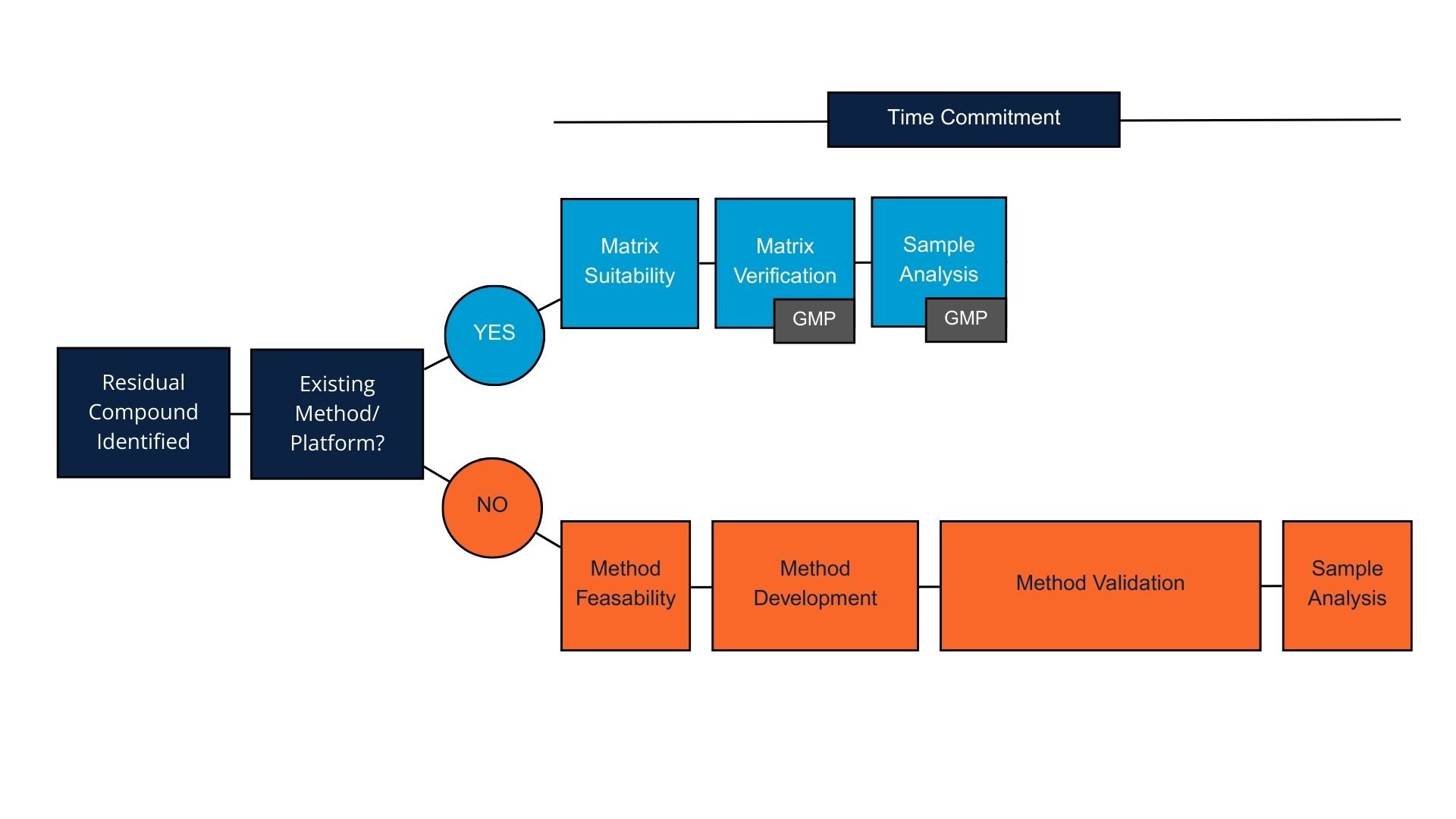
Figure 1. Biologic product platform methods decision tree
Proprietary manufacturing processes using novel reagents or unusual expression systems may generate impurities without established detection methods. These situations require custom analytical development using techniques ranging from advanced mass spectrometry to sophisticated bioassays.
Challenging product matrices also sometimes necessitate custom methods. Matrices containing extremely high protein concentrations, salt concentrations, preservatives, and surfactants are subjected to matrix interference and may require method modifications substantial enough to qualify as custom development.
Regulatory strategy occasionally drives the decision toward custom methods even when platform approaches exist. Some sponsors prefer product-specific method development to demonstrate thorough characterization and control.
Element's Process-Related Impurities Testing Capabilities
Element's biologics testing capabilities support comprehensive analytical strategies spanning platform method implementation and custom method development, followed by samples testing, or validation and samples testing. For common process-related impurities where platform methods provide value, Element offers matrix feasibility for host cell protein testing using ELISA-based approaches, residual DNA quantification via ddPCR methods, and other common upstream and downstream process impurities.
When programs require custom analytical approaches due to unique matrices, novel impurities, or specialized performance requirements, comprehensive method development capabilities ensure that analytical challenges don't become program constraints. This includes advanced chromatography, mass spectrometry, immunoassays, and bioanalytical techniques applied to challenging analytical problems.
Element's pharmaceutical testing laboratories maintain quality systems designed for regulatory compliance, supporting both early development testing and GMP validation studies for regulatory submissions. Flexible engagement models accommodate programs at different development stages, from research-grade testing through commercial release and stability programs.
Ready to discuss analytical testing strategies for your biologics program? Connect with our team to explore how platform methods and custom development can support your process-related impurities testing needs.
Related Industries
By Engaged Expert



Related Services

Biopharmaceuticals & Biologics Analysis Services
Our clients develop safe, efficacious products with confidence thanks to Element’s Biologics and Advanced Therapy Medicinal Products (ATMPS) testing services.
Extractables and Leachables Testing Services
Element provides tailored extractables and leachables testing (E&L) studies to ensure patient safety and compliance with regulatory requirements.

Cell Culture & Cell-Based Bioassay Services
Element provides expert cell-based bioassay services, including potency testing, method validation, and regulatory support, helping biopharmaceutical companies streamline drug development and approval.

Chemistry, Manufacturing and Controls (CMC) & CDMO Testing Services
Element provides specialized analytical testing services designed for CDMOs, offering flexible capacity, regulatory compliance, and advanced capabilities that enable successful client program delivery across all development phases.

Compendial Monograph and Pharmacopeial Testing
Element provides expert compendial testing services to help pharmaceutical and biotech companies meet global pharmacopeial standards, ensuring product quality, compliance, and market readiness.

Pharmaceutical Quality Control Testing
Element provides pharmaceutical quality control testing, covering raw materials, APIs, and finished products. Our expert analytical development and validation help meet regulatory requirements and support all phases of product development.